
On the Frustration to Predict Binding Affinities from Protein–Ligand Structures with Deep Neural Networks | Journal of Medicinal Chemistry (acs.org)
* Open Access 가 아니라 Abstract 만
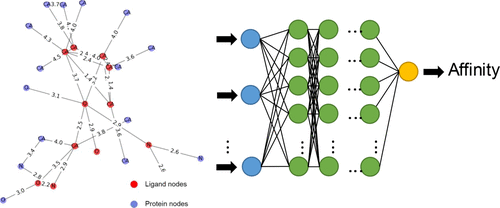
단백질 - 리간드 원자 스케일에서의 결합 강도는 drug discovery 의 초기 단계에서 아직도 큰 과제임.
이 논문에서는 modular message passing GNN 을 이용하여 ligand, protein 의 free, bound state 를 설명하려고 했고, 리간드-단백질의 비공유결합만으로는 설명이 어렵다는 것을 말하고 있다.
간단한 모델들도 training set 에 있는 근접한 protein 과 ligand의 결합력(결합 친화력) 을 잘 예측하는데, 이는 DNN 에 있어서 memorization 이 큰 역할을 하는 것으로 말할 수 있다.
이 연구에서는 다른 protein, ligand 의 원자적인 환경은 고려하지 않고 오직 비공유결합만 고려할것을 제시함.
Hidden biases 를 제거하는데에는 공동체의 노력과 좀 더 dense 한 protein-ligand matrice 가 필요할 것.
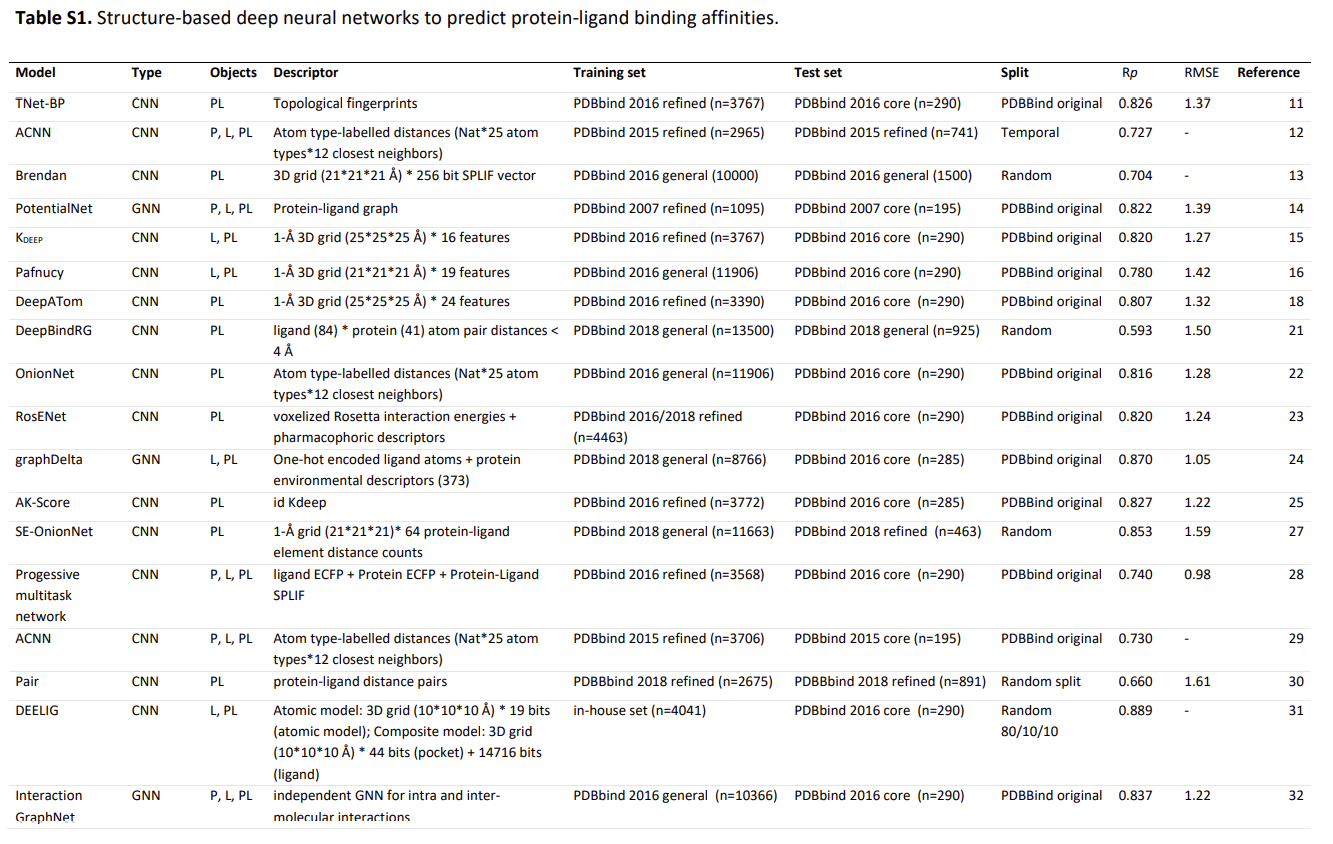
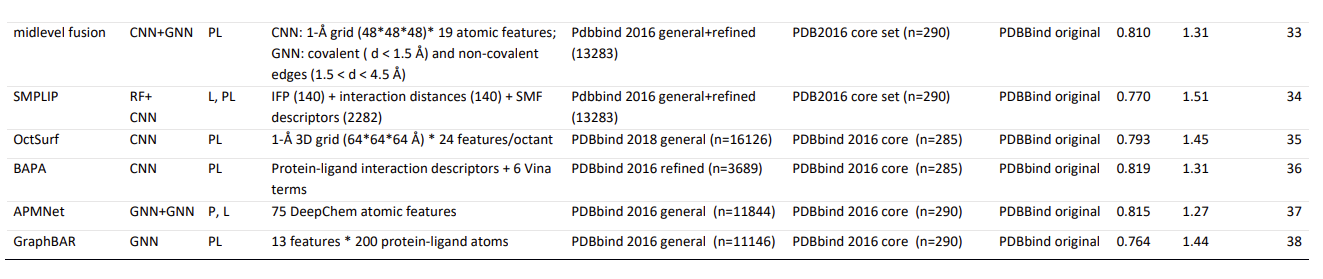
Supple 에 보니 Model, dataset 들이 잘 나와있어 첨부해놓음.
'논문 리뷰 > Abstract only' 카테고리의 다른 글
| The metastatic spread of breast cancer accelerates during sleep (0) | 2022.07.03 |
|---|---|
| Using deep learning to annotate the protein universe (0) | 2022.06.26 |
| Structural basis of GABA reuptake inhibition | Nature (0) | 2022.06.19 |
| People construct simplified mental representations to plan (0) | 2022.05.29 |
| A natural mutator allele shapes mutation spectrum variation in mice (0) | 2022.05.29 |
댓글