
Network-based machine learning approach to predict immunotherapy response in cancer patients | Nature Communications
Kunyoo Shin & Sanguk Kim Groups
Nature communications, 2022
Introduction
ICI 를 쓰는 것은 chemo 보다 부작용이 적고, 효과가 길게 감. 하지만 소수의 환자들만이 면역항암요법에 반응한다는 것이 한계임. 따라서 치료 전에 반응을 하는 환자군을 예측하기 위한 바이오마커를 알아내는 것이 중요함.
면역항암요법에 precision medicine 의 적용의 문제점 중 하나는 다른 암 환자군들에서 약물 반응을 예측하기 위해, 면역항암요법을 받은 환자들에서 marker 를 찾아내는 것임.
예를 들어, PD-L1 발현은 FDA 승인받은 CDx test 인데, 어떤 연구에서는 NSCLC 에서 PD-L1 발현과 ICI 반응성 사이에 큰 관련이 없다고 나왔는데 어떤 연구에서는 관련이 없다고 나왔음. 심지어 어떤 연구에서는 ICI reponder 에서 PD-L1 발현이 낮다고도 나온 연구가 있음.
최근의 많은 연구들은 ICI 치료를 받지 않은 암환자로부터 바이오마커를 찾기를 시도했고, 실제로도 성공했지만 이 접근법은 ICI 특이적인 바이오마커가 ICI 치료받지 않은 환자들로부터는 나오지 않을수도 있다는 한계점이 있음. 따라서 ICI치료를 받은 환자들로부터 바이오마커를 찾아낼 수 있는 방법론의 개발이 필요한 상태임.
이 연구에서는 기계학습 프레임워크를 활용해 ICI 데이터셋을 이용하여 정확한 예측과 바이오마커 탐색을 진행했음. 700샘플 이상의 암환자 유전자 발현량 데이터로 responder, non-responder 를 예측했으며, 이를 위해 PPI(Protein-protein interaction) 네트워크 안에 있는 면역항암제의 타겟과 가까이에 있는 생물학적 pathway 를 찾는 등의 네트워크 기반의 접근방식을 채택.
찾은 바이오마커의 검증을 위해 within-study cross validation, across-study prediction 을 진행함. 또한 TMB + 찾은 마커 를 같이 사용했을 때, overall survival 의 prediction 에서 더 좋은 예후를 보였음.
Results
이전 연구에서 항암제와 관련된 바이오마커는 ppi 상에서 약물 타겟과 가까이 위치한다는 것을 밝혔음. 이를 이용하여 ICI 타겟과 가까이 있는, ICI 반응과 연관되어있는 생물학적 pathway 를 찾으려고 했음.

STRING DB 를 사용하였고 ( STRING 에서 제공하는 score 가 있는데, 700 이상인 interaction 만 사용함 ) , 16,597 개 node, 420,381개 edge 를 사용함.
Network propagation 을 이용한 identification
- ICI 타겟을 이용한 network propagation.
- ICI 타겟을 seed gene 으로 해서 ICI 타겟의 influence 를 퍼트리기 위해서 진행.
- ICI 타겟에 가까운 node 일수록 influence score 가 높고, 이에 기반하여 top 200 유전자를 뽑은 후 Reactome pathway 에서 enrich 된 pathway 를 찾음. 그 후 pathway 에 기반하여 면역항암제 반응 예측 진행.
- 위에서 찾은 pathway 를 Network-Based Biomarkers (NetBio) 라고 명명함.
면역항암제 예후예측을 진행하기 위해, NetBio 를 input 으로 사용함. Negative control 로는 gene-based biomarker 를 포함한 여러 관련 유전자 / pathway 를 사용함.
| Negative control (1) from Supplmentary figure | Negative control (2) from Supplementary figure |
 |
 |
Input feature 의 발현량을 사용하여 , Logistic regression 으로 ML 모델 생성.
Prediction ( on prediction of drug response )
- Immunotherapy 후에 줄어든 tumor 의 size
- Survival
을 예측하는 것을 목표로 함
Datasets
- Within-study prediction
Train, test 가 한 코호트에서.
LOOCV, Monte-Carlo CV
-> 여기서의 Monte-Carlo CV 라는 것은 train : tes = 8 : 2 로 random sampling 하는 과정.
-> 이것을 한 이유는 적은 수의 training set 으로도 robust 한 성능이 나온다는 것을 보여주기 위함.
-> 72번 시행 중 70번이 다른 바이오마커보다 좋은 성능을 냈음. ( student t test )
- Across-study prediction
두 개의 독립된 데이터셋을 train, test 로 나누어서 사용
적은 수, 많은 수의 dataset 을 반복적으로 사용함으로서 model 의 robustness 를 보여주고자 함.
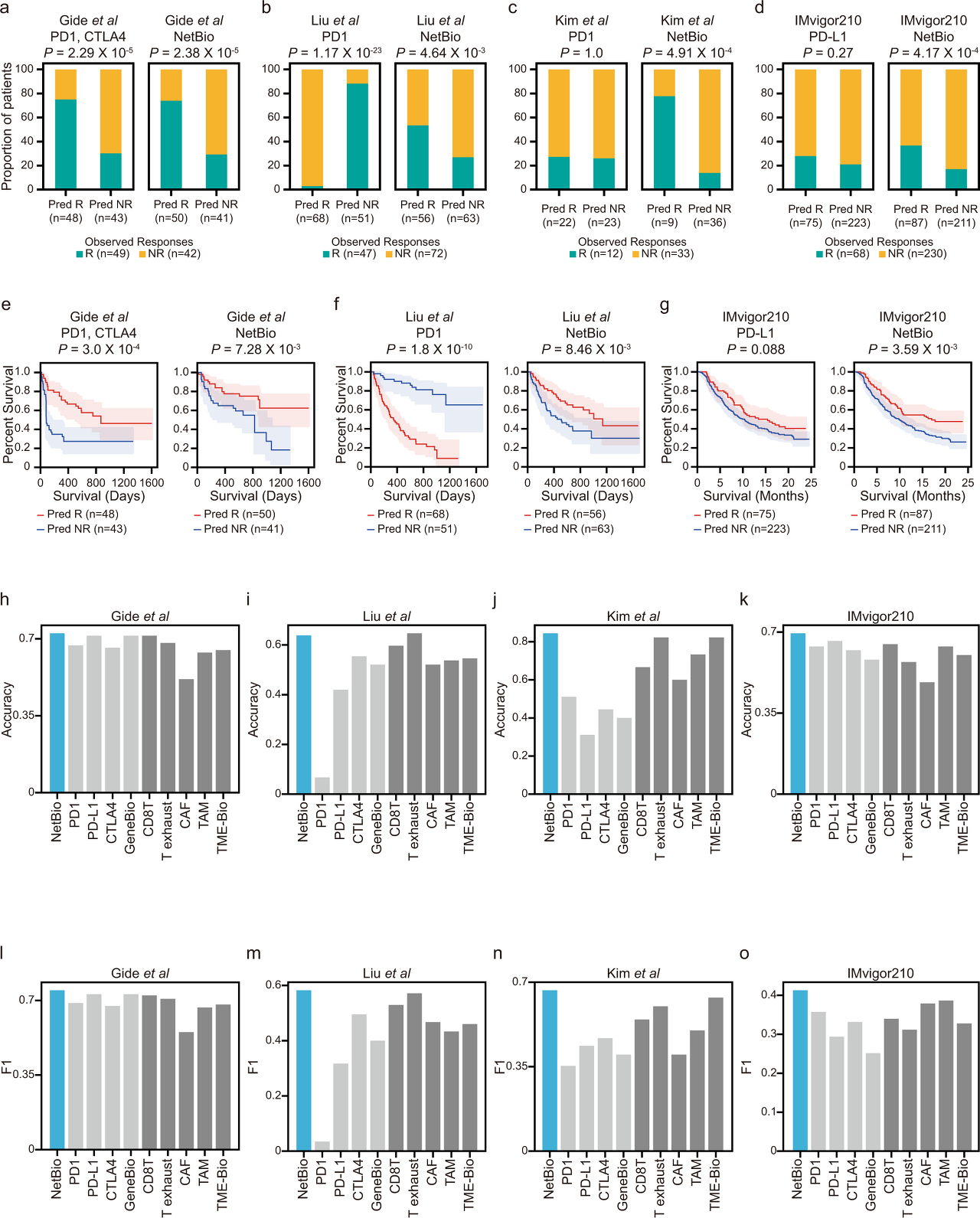
이 그림에서는 기존 마커들과의 비교를 하고 있음.
처음 피규어에서는 PD-L1, CTLA4 와의 비교를 통해 drug target 과 NetBio 의 비교를 진행,
두 번째에서는 overall survival 과의 비교, supple 에서는 PFS 와도 비교 진행.
결과적으로는 NetBio 를 이용하여 예측한것이 더 성능이 좋았다는 점이고, Liu cohort 에서는 기존의 결과와 다르게 예측되었다는 것을 말하고 있음.
H-O 까지의 figure 는 기존의 ICI 관련이라고 알려진 biomarker 들에 대한 것을 진행하고 있음.
 |
 |
| Figure from original paper ( Supplementary Figure ) Monte carlo cross validaton ( Random sampling, 100 iteration 해서 비교함 ) |
Figure from original paper ( Supplementary Figure ) Clinical setting 에서 사용되는 점수인 TPS 로 예측을 진행했을때와, NetBio 로 예측을 진행했을 때의 비교. Clincal setting 에서 사용하는 점수로 예측한 결과보다 좋은 성능을 나타낸다. |
ML 모델의 적용점 중 하나인 일반화가 가능한가에 대해 테스트하려고 3개의 독립적인 melanoma cohort 로 test진행함.
AUPRC 로도 했는데,
이 때 3개 코호트 각각보다 높다는 것을 보여주고, 합쳐진 코호트에서도 높다는 것을 보여줌.
 |
 |
| Original figure from paper 3개 데이터셋보다 각각 높다는 것을 보여줌 |
Original figure from paper 3 개 데이터셋을 합쳐서 예측한 것 보다 높다는 것을 보여줌 |
뒤에서는 A cohort에서 train 한 후 B cohort 에서 test 한 것도 결과가 잘 나왔다고 말하고 있고,
이렇게 다양하게 시도를 해 보는 이유는 dataset 관계없이 예측을 잘 한다는 것.
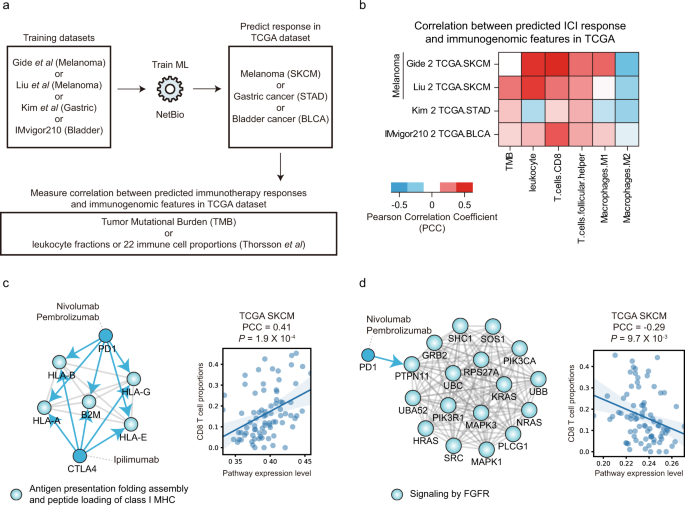
다음으로는 Immunotherapy 의 결과를 예측하는 것이 주된 목적이므로, NetBio 가 immune microenvironment 를 잘 설명할 수 있는가를 보기 위해, TCGA 를 포함하는 데이터셋에서 immune contexture와 예측된 결과의 correlation 을 보고자 했음. ( 예측이 잘 되면 correlation 도 좋아야 하므로.)
immune contexture 는 이전에 다른 그룹에서 출판된 논문에서 나온 immunogenic scores 를 사용. 결과적으로는 잘 나왔고, 암종을 섞어서 했을 때는 correaltion 이 유의하지 않게 되었다 함. ( Immune mircoenvironment 의 cancer-specificness )
이를 예측하는 데 있어서 어떤 NetBio pathway 가 가장 중요했는지 알아보기 위해서 Feature importance 를 사용하여 top 10 pathway 를 뽑아봄. (여기서부터 pathway level 의 설명이 시작됨)
antigen presentation folding assembly and peptide loading of class I MHC
FGFR signaling
이런 식의 pathway 가 나왔다고 하고, 이 pathway 들을 이용해서 possible biological mechanism 제시함.
이 다음으로는 NetBio pathway 의 expression level 이 immune context 와 연관되어있는 것에 대한 설명이 이어짐.
Curate 된 pathway 의 expression 과 , immune phenotypes 의 비교를 진행한 것 인데, immune phenotype 간에 pathway 수준에서의 expression level 을 보고함.
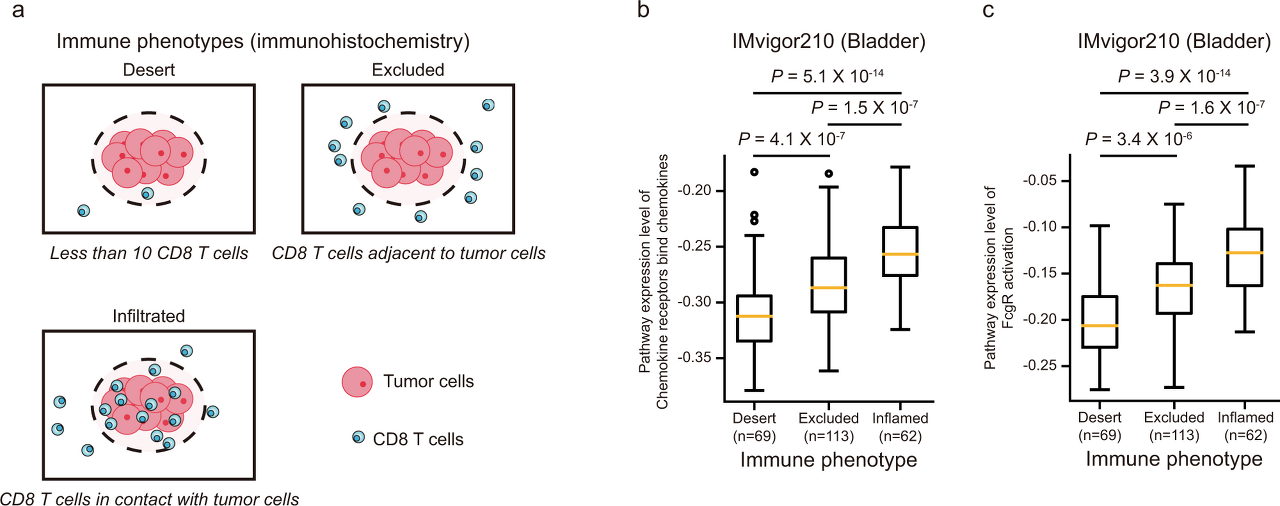
마지막으로는 TMB level 와 NetBio 를 합쳐서 예측하는 것을 진행함. 이는 높은 TMB level 이 ICI 치료의 benefit 과 연관되어 있는데 반해, responder 와 non-responder 사이에서의 TMB level 이 비슷한 경우가 많이 때문임.
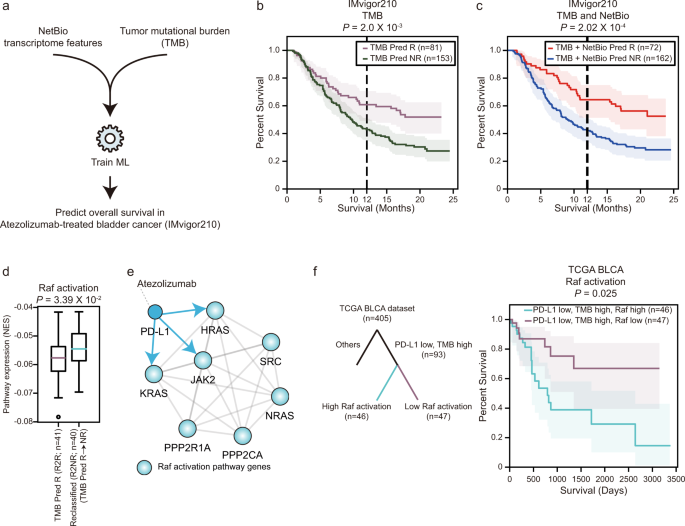
확실히 TMB 를 같이 썼을 때 survival 예측이 좋아진 것이 보임.
이 결과에 대해서, TMB alone 으로 예측했을 때 보다 같이 예측했을 때 Responder -> Non-responder 로 간 case ( R2NR ) 과 반대로 간 케이스가 있었는데, 이들의 검증에서 NR 로 바뀐 환자들은 reponder group 과 비교했을 때 overall survival 이 감소되었고, R 로 바뀐 환자들은 overall survival 이 길었음.
이렇게 성능이 좋아진 이유 ( reclassifiy 가 잘 된 이유) 를 찾으려고 했는데,
reclassifiy 가 된 그룹에서 TMB level 은 비슷하다는 것을 보였음. 따라서 TMB level 은 confounding factor 가 아니라고 여겨짐.
그렇다면 관여된 transcriptomic feature 를 찾으려고 했는데,RAF activation pathway 가 그것이었음. PPI 상에서 봤을 때도 , 관련된 유전자들을 볼 때 이들은 PD-L1 의 direct neighbor ( 거리가 1 인 gene 을 말하는 듯 ) 이어서, 굉장히 consistent 했음.
이렇게 찾아진 RAF activation pathway 가 과연 ICI 치료의 biomarker 로서 사용될 수 있는가 ? 를 검증하기 위해, PD-L1 발현, TMB, RAF 의 발현량을 overall survival 과 연관성 조사를 진행하였음.
- PD-L1 발현량이 낮을 때 ( PD-L1 inhibition 상황에서 )
- TMB level 이 높을 때
이렇게 두 가지의 상황을 가정하고 이 상황에서 RAF 의 활성화가 overall survival 에 영향을 미치는가 ? 에 대한 검증을 진행함.
결과적으로는 low PD-L1, high-TMB 에서 RAF activation pathway 가 overall survival 에 통계적으로 의미있는 영향을 미쳤음. RAF pathway 가 activation 되면 overall survival 이 낮아졌음. 이는 PD-L1 저해제 치료를 받은 환자들에서 치료에 대한 면역이 생긴다는 finding 과 일치하였음.
Discussion
STRING score 900 으로도 해보았는데, 대부분의 pathway 는 기존 모델 (Score 700) 과 비슷했으나 성능이 조금 떨어지는 부분이 있었음 : 이는 작은 네트워크가 중요하다는 것을 말함.
그럼에도 불구하고 다른 marker 들과 비교했을 때 900 모델도 더 낫거나 같은 성능을 보였음.
Methods
NetBio detection
2단계로 이루어짐
1) ICI 타겟과 가까이에 있는 gene들 발굴
- NetworkX 의 pagerank 를 사용하여 발굴함
- Pagerank parameter : ICI target=0, 모든 다른 gene =1, 다른 parameter 는 default
- Influence score 가 높은 top 200 gene 발굴
2) 1) 의 유전자들에 enrich 되어있는 pathway 발굴
- GSEA 돌려서 target 과 근접한 유전자가 얼마나 많이 enrich되어있는지 계산
- 이는 Hypergeomeric test 으로 pval 확인
- 472,323,292,353 pathway 가 선택됨
- pathway 의 expression 을 사용하여 ML model train
이 과정에서 NetBio pathway 의 발현이 treatment 전, 진행 중 과 비슷하다는 것을 발견했는데, 여기서 differentially expressed pathways (DEPs) 를 발굴하여 비교하였음. 결과적으로는 두 그룹 간 DEP 가 발견되지 않아서, 이를 통해 NetBio의 발현 profile 은 치료 중에도 변하지 않는 다는 것을 발견.
Transcriptome similarity
- 각 환자마다 다른 코호트와 비교해서 spearman rank correlation 을 계산함
- spearman correaltion 에서 제일 큰 값을 가져옴
- 1,2, 번째를 모든 환자 쌍 ( 두 코호트 ) 에 대해서 계산함.
결과적으로 두 코호트에서 나온 값을 통계처리.
* Pathway 가 300여개 선택이 되어서, 이것이 기계학습의 input 으로 들어가는 것 인데, 그렇다면 pathway 의 gene 이 겹치는 경우도 있으니까 pathway 자체의 representative expression value 를 사용한 것 일까 ?
-> We used the normalized enrichment score (NES) to estimate the pathway expression levels of each sample (Supplementary Data S7)
-> 결국에는 Enrichment scroe 를 사용하여 machine learning 의 input 으로 들어간 것.
그렇다면 총 feature 가 300여개이고, sample 은 700여개로 충분하니 학습이 잘 될것.
-> 하지만 이 경우 gene - level 의 feature importance 는 보기 어렵다. 상당히 macro 한 모델.
처음 figure 를 봤을 때에는 node 기준의 classifier 로 생각했는데, 물론 어느 측면에서는 그것도 맞지만 cluster 를 진행한 후 그 cluster 를 대표하는 representative value 를 구하여 기계학습에 적용한 경우.
이전에 읽었던 Nature 논문인 Biologically informed deep neural network for prostate cancer discovery | Nature
과 비슷한 느낌의 논문이었지만, 여러가지 validation / test cohort 를 이용함으로서 모델을 만드는데 있어 신뢰감을 준다.
댓글